> For the complete documentation index, see [llms.txt](https://knowledge.illumina.com/llms.txt). Markdown versions of documentation pages are available by appending `.md` to page URLs; this page is available as [Markdown](https://knowledge.illumina.com/software/cloud-software/software-cloud-software-reference_material-list/000007134.md).
# 使用BaseSpace基因云计算平台和bcl2fastq2 2.17以及以上版本软件时,如何排查拆分异常情况
\
本技术文档适用于[BaseSpace基因云计算平台](https://basespace.illumina.com/)和[bcl2fastq2](http://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html) 2.17以及以上版本软件的数据拆分问题。
BaseSpace基因云计算平台和bcl2fastq2 v2.17+在数据拆分结束后均会产生拆分总结文件,即DemuxSummaryF1L#.txt。其中,L#代表的是流动槽(Flow Cell)上的通道(Lane)编号,并且针对每条Lane都会产生一个对应的文件。该文件可以用于排查拆分异常情况的原因。
**在哪里可以看到DemuxSummaryF1L#.txt**
***bcl2fastq2 2.17以及以上版本软件***
FASAQ文件生成结束后,在stats文件夹中会产生DemuxSummaryF1L#.txt文件。Stats文件夹位于指定的输出目录中。
***BaseSpace基因云计算平台***
针对所有的测序运行runs,在FASAQ文件生成结束后,系统会把DemuxSummaryF1L#.txt存储在相应的project中。用户可以通过点击project中分析中的FASTQ Generation链接,在弹出的Summary界面点击View Files(图1),就可以在文件列表中找到DemuxSummaryF1L#.txt文件。
1. 进入 **Run** 页面,在 **“Latest Analyses”** 下选择 **FASTQ Generation** 链接(见图 1)。

**图 1:**列出 FASTQ Generation 分析的 Run Summary 页面
2\. 在 **Analysis Info** 页面中,向下滚动到 **Log Files**(见图 2)。你可以在日志文件列表中找到 **DemuxSummaryF1L#.txt** 文件。

**图 2:**Analysis 页面中的日志文件列表
**DemuxSummaryF1L#.txt含有哪些内容?**
DemuxSummaryF1L#.txt含有两个部分。正如图3显示的,文件的上半部分是一个tab分割的表格,该表格总结了每个tile的样本拆分情况。表格的左侧是流动槽上的tile列表。 在表格的顶部,样品按输入样品表的顺序列出。 样品0指的是未拆分的reads。 下表显示每个tiles上reads拆分到每个样本的百分比。 通常,在所有tiles上针对某一个样本的拆分比例应该是接近的。用户可以利用tile的总结信息明确与特定tile有关的拆分问题。
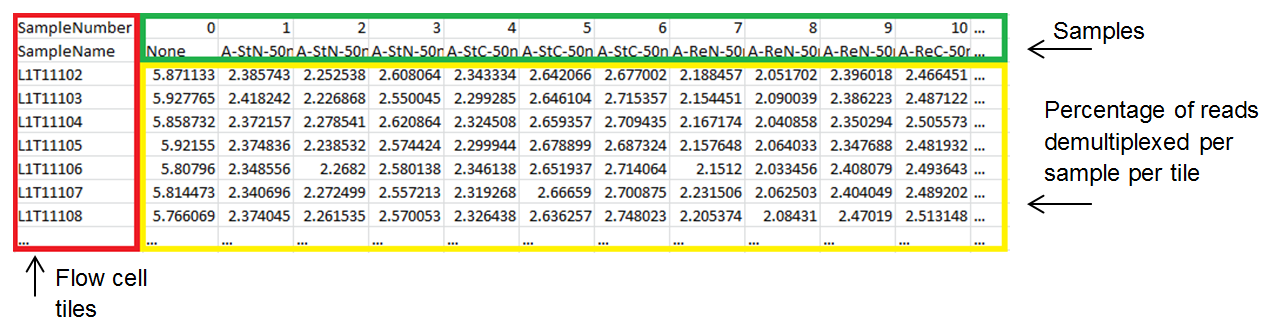
**图 3:Tile summary 部分显示了按 tile 统计拆分到各个样本的 reads 百分比。**
DemuxSummaryF1L#.txt文件(图4)的第二部分列出了前1000个比较多的未拆分的index序列以及对应的数目(或者说分配到每个index序列的cluster的数目)

**图4:DemuxSummaryF1L#.txt文件中index序列部分截图,该部分列出了前1000个没有拆分到样本的index序列**
当遇到拆分问题时,可以使用此列表将预期index序列(SampleSheet中)和测序测到的实际序列进行比较。
这些列表可以揭示一些低拆分比例的常见原因:
* 在样本表(SampleSheet.csv)输入了错误方向的index序列
* 在样本表中输入了错误的index(比如Nextera vs TruSeq UD或者index A001 vs index A006)
* 不同lane的样本发生了混合
* index测序质量差
* 序列中含有Ns,N是指碱基检出软件不能识别该位置的碱基
* 对于[单通道](https://www.illumina.com/content/dam/illumina-marketing/documents/products/techspotlights/cmos-tech-note-770-2013-054.pdf)测序仪器(iSeq)和[双通道](https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology/2-channel-sbs.html) (MiniSeq,NextSeq 500/550以及NovaSeq),Poly-G序列说明没有读取到index序列。poly-G序列是典型的Phix序列,这是因为Phix序列是没有index的。
\
\
\
| |
| :-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------: |
| *For any feedback or questions regarding this article (Illumina Knowledge Article #7134), contact Illumina Technical Support* [*techsupport@illumina.com*](mailto:techsupport@illumina.com?subject=Question%2FFeedback%20Regarding%20Illumina%20Knowledge%20Article%20#000007134%20-%20Software%20\&body=Dear%20Illumina%20Technical%20Support,%0D%0A%0D%0A)*.* |
---
# Agent Instructions
This documentation is published with GitBook. GitBook is the documentation platform designed so that both humans and AI agents can read, navigate, and reason over technical content effectively. Learn more at gitbook.com.
## Querying This Documentation
If you need additional information that is not directly available in this page, you can query the documentation dynamically by asking a question.
Perform an HTTP GET request on the current page URL with the `ask` query parameter, and the optional `goal` query parameter:
```
GET https://knowledge.illumina.com/software/cloud-software/software-cloud-software-reference_material-list/000007134.md?ask=&goal=
```
`ask` is the immediate question: it should be specific, self-contained, and written in natural language.
`goal` is optional and describes the broader end goal you are ultimately trying to accomplish on behalf of the user. GitBook uses it to tailor the answer towards what is most useful for that goal.
The response will contain a direct answer to the question and relevant excerpts and sources from the documentation.
Use this mechanism when the answer is not explicitly present in the current page, you need clarification or additional context, or you want to retrieve related documentation sections.